






About Us
Executive Editor:Publishing house "Academy of Natural History"
Editorial Board:
Asgarov S. (Azerbaijan), Alakbarov M. (Azerbaijan), Aliev Z. (Azerbaijan), Babayev N. (Uzbekistan), Chiladze G. (Georgia), Datskovsky I. (Israel), Garbuz I. (Moldova), Gleizer S. (Germany), Ershina A. (Kazakhstan), Kobzev D. (Switzerland), Kohl O. (Germany), Ktshanyan M. (Armenia), Lande D. (Ukraine), Ledvanov M. (Russia), Makats V. (Ukraine), Miletic L. (Serbia), Moskovkin V. (Ukraine), Murzagaliyeva A. (Kazakhstan), Novikov A. (Ukraine), Rahimov R. (Uzbekistan), Romanchuk A. (Ukraine), Shamshiev B. (Kyrgyzstan), Usheva M. (Bulgaria), Vasileva M. (Bulgar).


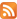
Materials of the conference "EDUCATION AND SCIENCE WITHOUT BORDERS"
 PDF
PDFIn the study of liquid crystals, the focus is mainly on the study of phase transitions [1], including the formation of a one-dimensional and two-dimensional structure. Phase transitions in liquid crystals are primarily determined by intermolecular interactions: the side in the plane and at the ends of the molecular pairs. These intermolecular interactions determine the physical properties of liquid crystals as well as the type of physical and chemical processes occurring in them.
Substituted phenylpropargyloxybenzenes (PPOB) [2] with the general formula XC6H4OR (X ─ polar substituent, R=CH2CCC6H5) are nematic liquid crystal compounds. According to the nature and position of substituent X in the aryl ring of phenoxy fragment the melting point and thermal stability of these substances change. These and other physical properties of liquid crystals (LC) are due to the interaction of mesogenic molecules. In turn, molecular interactions play an important role in determining the molecular conformations of LC. In this paper, the computer simulation of molecular pairs of thermotropic nematic liquid crystal (fenylpropargyloxybenzene) by quantum-chemical method RHF/6-31G(d,p) is considered.
The purpose of this work was to determine the most characteristic behavior of molecules surrounding a fixed molecule and an influence of the internal rotation on the energy of complex.
For stacking interactions (side) a distance of 6 Å (to avoid the van der Waals interactions) is selected:
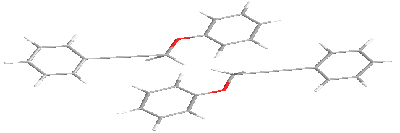
For in-plane
and terminal interactions
 intermolecular distance equals to 8 Å.
intermolecular distance equals to 8 Å.
Variants of the antiparallel stacking of the molecules as the most characteristic for polar liquid crystals were considered. One of the molecules was assumed fixed. When stacking interactions in molecular pair one of the molecules is fixed in the plane, while the second is located along the Z axis at a distance of 6 Å relative to the first molecule. When interacting in the plane interacting molecules located along the Y axis at a distance of 8 Å with respect to the fixed molecule. There is chosen as the origin atom being next to the center of mass of the molecule. The X-axis is directed along a line located parallel to the long axis of the molecule, the Y axis is in the plane of the molecule, and the Z axis is perpendicular to the X-Y. Next, we consider possible options for the relative position of a pair of molecules fenylpropargyloxybenzenes for stacking, plane and terminal interactions. For these types of interactions the potential energy surface in the coordinates of energy - torsion angles is calculated.
Using the computer simulation of molecular interactions of thermotropic PPOB, we held a scanning the potential energy surface by two torsion angles corresponding to rotations of the benzene rings about the carbon-oxygen and carbon-carbon bonds. For the dimers the angle between the long axes of the molecules is ~ 0, i.e. molecules in the short-range order are strictly parallel. One of two identical molecules taken at rest, and the center of gravity position and orientation of the second molecule were varied relative to the first one. The moved molecule was used as a rigid rotor model, as it is well-known that the intermolecular forces have mainly an effect on the angles of internal rotation, almost without changing the bond lengths and bond angles.
The quantum chemical calculations were performed in the single-determinant Hartree-Fock approximation using a split-valence basis 6-31G(d,p). We have simulated and studied with electron-donating, electron-withdrawing substituents in the para-position of aryl ring of phenoxy fragment and reference unsubstituted PPOB. Below the potential energy surface of the dimer of fenylpropargyloxybenzene (X-axis (f1)) is demonstrated.
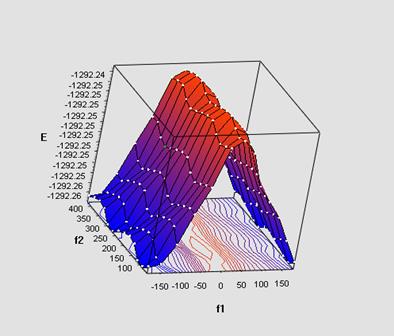
The calculations show a marked change in the energy of complex at the rotation of p, π - conjugated phenoxy fragment and a weak influence of the internal rotation of the phenyl ring.
2. S.A. Shchelkunov, L.K. Abulyaisova, S.O. Mataeva, et al, “Paradoxes in thermal stability of the liquid-crystal phase of phenylpropargyl phenyl ethers: I. Synthesis and mesogenic properties of phenylpropargyl phenyl ethers”, Russian Journal of General Chemistry, 2001, Vol. 71, No. 7, P. 1130-1132.
Abulyaissova L.K., S.O. Kenzhetayeva, M.T. Alimbayeva, L.Ph. Dyusembayeva COMPUTER SIMULATION OF MOLECULAR INTERACTIONS OF NEMATIC
PHENYLPROPARGYLOXYBENZENES
. International Journal Of Applied And Fundamental Research. – 2013. – № 2 –
URL: www.science-sd.com/455-24241 (22.07.2026).